Abstract
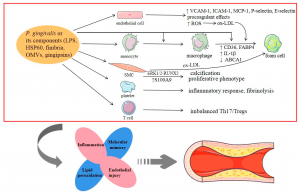
Porphyromonas gingivalis (P. gingivalis) is a periodontal pathogen that can accumulate with other organisms in subgingival plaque biofilms and is associated with periodontal disease. P. gingivalis fimbriae (FimA) is a filamentous structure on the surface of bacteria that is closely associated with bacterial adhesion and colonization of host tissues and plays an essential role in biofilm formation. The present study aimed to construct prokaryotic P. gingivalis FimA expression plasmids, purify a FimA fusion protein, and explore the effect of a recombinant FimA protein on the inflammatory response in human peripheral blood mononuclear cells (PBMC).
Porphyromonas gingivalis Recombinants FimA prokaryotic expression plasmids were constructed using gene cloning and recombination technology. SDS-PAGE was used to evaluate purified recombinant FimA protein. Cell proliferation rate and inflammatory cytokine expression of PBMC treated with FimA fusion protein with or without toll-like receptor 4 (TLR4) small interfering (si) RNA transfection were detected by CCK-8 assays and ELISA, respectively. Expression levels of TLR4, nuclear factor kappa light chain enhancer of activated B cells (NF-κB), and primary myeloid differentiation response 88 (MyD88) in PBMC were detected by Western blot analysis and quantitative polymerase chain reaction with reverse transcription.
A high purity FimA fusion protein was obtained. FimA fusion protein treatment significantly increased PBMC proliferation and promoted the release of tumour necrosis factor-α (TNF-α), interleukin (IL)-6, matrix metalloproteinase (MMP)-8, and MMP-9 in PBMC. TLR4 interference reversed the effects of FimA fusion protein on PBMC proliferation and inflammatory cytokine release. The expression levels of TLR4, NF-κB and MyD88 in PBMCs were significantly increased after FimA fusion protein treatment, whereas the expression levels of these genes at mRNA and protein levels were significantly decreased in PBMCs after treatment. FimA fusion protein treatment and TLR4 interference.
Materials and methods
- Subject Recruitment, Blood Sampling, and PBMC Culture
A total of 12 healthy volunteers in good periodontal health (37.5 ± 6.3 years) with no known periodontal pocket formation or attachment loss, no alveolar bone resorption, and no systemic disease were recruited from the Second Affiliated Hospital of the Medical University of Jinzhou from September 2017 to December 2017. Additional exclusion criteria were the presence of gingivitis, periodontal disease, orthodontic treatment, or a history of long-term use of antibiotics or other medications. All subjects who participated in the experiment underwent a periodontal examination by the same dentist. Subjects clearly understood the purpose of the experiment, agreed to participate and gave written informed consent.
Specimen collection was approved by the Ethics Committee of the Second Affiliated Hospital of Jinzhou Medical University (Jinzhou, China). A total of 10 ml of venous blood was collected from each volunteer and stored in tubes with EDTA anticoagulant (BD Biosciences, San José, CA, USA). PBMCs were isolated by the Ficoll-Paque density gradient centrifugation method (650 × g; 18 °C for 20 min) and cultured in RPMI-1640 medium (Thermo Fisher Scientific, Inc., Waltham, MA, EE). bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc.) All cells were grown in an incubator containing 5% CO2 at 37°C with 100% humidity.
- P. gingivalis culture and genome extraction
A frozen liquid culture (-80°C) of P. gingivalis 33277 strain [American Type Culture Collection (ATCC), Manassas, VA, USA] was thawed at room temperature and inoculated into a Petri dish containing brain heart infusion (BHI) agar with 1 mg/l vitamin K, 1 g/l yeast extract and 5 mg/l hemins (China Medical University, Shenyang, China). Cells were grown under anaerobic conditions at 37°C for 4 days. Surface colonies were scraped with sterile loops onto a sterile ultraclean bench and inoculated into BHI liquid culture medium.
Cells were cultured for ~24 h until bacterial growth reached the log phase. Then, the supernatant was discarded after centrifugation at 4000 x g for 15 min at 4 °C to obtain the bacteria, and P. gingivalis genomic DNA was extracted. Genomic DNA was extracted using the Wizard Genomic DNA Purification Kit (Promega Corporation, Madison, WI, USA), following the manufacturer’s instructions.
- Semi-quantitative polymerase chain reaction (PCR) amplification of target genes
Based on the FimA sequence of P. gingivalis ATCC 33277 available in the National Center for Biotechnology Information GenBank database, primers (forward, 5′- ATT AGG ATC CAT GGT GGT ATT GAA GAC CAG C-3′; reverse, 5′-ATA TCT CGA GCC AAG TAG CAT TCT GAC CAA CGA G-3′) were designed using Premier 5.0 software (Premier Biosoft International, Palo Alto, CA, USA) to remove the stop codon from the original sequence and add a start codon. Primers were synthesized by Nanjing GenScript Biotechnology Corporation (Nanjing, China).
The FimA gene was amplified by the LA Taq enzyme (Takara Biotechnology Co., Ltd., Dalian, China). Cycling conditions included 94 °C for 5 min, followed by 25 cycles of 94 °C for 30 s, 58 °C for 30 s, and 72 °C for 2 min, with a final extension at 72 °C for 10 min. The PCR products were identified by 1% agarose gel electrophoresis in tris acetate buffer (40 mM tris acetate, 1 mM EDTA, pH 8.0). Products were visualized with ethidium bromide by UV transillumination.
- RNA interference assay
Shanghai GenePharma Co., Ltd., (Shanghai, China) synthesized small interfering RNAs (si) targeting toll-like receptor 4 (si-TLR4) and negative siRNA with a random sequence. The target sequences for TLR4 were: Sense, 5′-GGG CUU AGA ACA ACU AGA ATT-3′; and antisense, 5′-UUC UAG UUG UUC UAA GCC CTT-3′, and the sequences for the negative siRNA were: Sense, 5′-UUC UCC GAA CGU GUC ACG UTT-3′; and antisense, 5′-ACG UGA CAC GUU CGG AGA ATT-3′. The concentration used was 20 nM. All plasmids and oligonucleotides were transfected using Lipofectamine® 2000 (Invitrogen; Thermo Fisher Scientific, Inc.) according to the manufacturer’s instructions. After 48 h, cells were harvested and subjected to further experimentation.
- Cell proliferation assay
PBMC (1 × 103 cells/well) were seeded in 96-well plates and treated with FimA fusion protein (2, 4, and 6 μg/ml) with or without siTLR4 transfection at 37 °C and 5% CO2 for 12, 24 and 48 hours. Cell proliferation was detected using a Cell Counting Kit-8 (CCK-8; Dojindo Molecular Technologies, Inc., Kumamoto, Japan). An aliquot of 10 μl of CCK-8 solution was added to the treated PBMCs and incubated at 37 °C for an additional 4 h. Cells were washed twice with PBS and then incubated at 37°C for 1-4 h. The absorbance value at 450 nm of each well was measured with a 96-well plate reader (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
- ELISE
PBMCs were seeded uniformly in 6 wells at a density of 1 × 106 cells/mL and treated with FimA fusion protein (2, 4, and 6 μg/mL) with or without transfection with siTLR4 at 37 °C and 5 %. CO2 for 12, 24 and 48 h. Cell culture media from all groups were collected and centrifuged at 8,000 × g for 20 min at 4 °C to remove debris.
The supernatant (400 μl) was collected from PBMCs to measure concentrations of TNF-α, IL-6, MMP-8, and MMP-9 with an ELISA-based capture assay using commercial TNF-α (cat. no. PT518) and IL-6 ELISA Kits (Cat. No. PI330) (Beyotime Institute of Biotechnology, Haimen, China), and MMP-8 (Cat. No. DMP800B) and MMP-9 ELISA Kits ( Cat. No. DMP900) (R&D Systems, Inc., Minneapolis, MN, USA) according to the manufacturer’s instructions. The absorption was measured at 450 nm. The concentration of inflammatory cytokines was determined by comparing the relative absorbance of the samples with the standards.
- Quantitative reverse transcription PCR (RT-qPCR)
Total RNA was extracted from PBMC treated with FimA fusion protein with or without siTLR4 transfection using TRIzol® reagent (Thermo Fisher Scientific, Inc.). The extracted RNA was then transcribed into double-stranded cDNA using a commercial reverse transcription kit (Invitrogen; Thermo Fisher Scientific, Inc.).
The cDNA was amplified using the following primers: TLR4 forward, 5′-CCG CTT TCA CTT CCT CTC AC-3′; reverse TLR4, 5′-CAT CCT GGC ATC ATC CTC AC-3′; nuclear factor of activated B cells (NF-κB) kappa light chain enhancer forward, 5′-CTT GCT TAG TTG GTC CTC-3′; reverse NF-κB, 5′-ACC CGA AGA GAA ACG A-3′; myeloid differentiation primary response 88 (MyD88) forward, 5′-AGA TGG ACC TCG GGA G-3′; reverse MyD88, 5′-ATC AAT CAC GCA CGA TTT-3′; β-direct actin, 5′-TCC CTG TAT GCC TCT G-3′; β-reverse actin, 5′-ATG TCA CGC ACG ATT-3′. The reaction system contained cDNA template (1 μl), primers (1 μl), 2X SYBR Green Mix (10 μl; Shanghai GeneCore BioTechnologies Co., Ltd., Shanghai, China), and RNase-free water (8 μl), with a total volume of 20 μl.
PCR was performed using the ABI 7500 system platform (Applied Biosystems; Thermo Fischer Scientific, Inc.). The 2−ΔΔCq method was used to calculate the relative gene expression of TLR4, NF-κB and MyD88 normalized to β-actin (20). PCR amplification was repeated in triplicate for each gene.
- Western blot analysis
Total protein was extracted from PBMC treated with FimA fusion protein with or without siTLR4 transfection by radioimmunoprecipitation assay (Beyotime Institute of Biotechnology). Protein concentration was evaluated by the BCA protein assay kit (Beyotime Institute of Biotechnology). Total lysates (50 mg) were resolved by 10% SDS-PAGE (Beyotime Institute of Biotechnology), followed by blocking for 1 h at room temperature in blocking buffer (cat. no. P0023B; Beyotime Institute of Biotechnology ).
Membranes were incubated with primary antibodies (rabbit anti-TLR4, 1:500 dilution, cat. no. ab13556; rabbit anti-NF-κB, 1:500 dilution, cat. no. ab207297; anti-rabbit Rabbit MyD88, 1:500 dilution, Catalog No. ab2064; rabbit anti-β-actin, 1:1000 dilution, Catalog No. ab8227; Abcam, Cambridge, UK). After rinsing with TBST, the secondary goat anti-rabbit horseradish peroxidase-conjugated antibody (1:5000 dilution; cat. no. ab6721; Abcam, Cambridge, UK) was added and the membranes were incubated at room temperature. environment for an additional 2 h. Image J software version 1.48 (National Institutes of Health, Bethesda, MD, USA) was used to analyze the relative density of the protein band.
- Statistic analysis
The statistical software SPSS 17.0 (SPPS Software, Inc., Chicago, IL, USA) was used for data analysis. Data are expressed as mean ± standard deviation. All experiments were repeated at least three times. Comparisons between two groups were analyzed using an independent samples t-test, while comparisons between multiple groups were analyzed using a one-way analysis of variance followed by Tukey’s honestly significant difference test. P<0.05 was considered to indicate a statistically significant difference.